About
Welcome to my homepage! My name is Haoyu Zhang, and I am currently an Earl Stadtman tenure-track investigator at National Cancer Institute, Division of Cancer Epidemiology and Genetics. My lab focuses on developing scalable statistical methods and software to analyze large-scale multi-ancestry genetic data to address questions related to health disparities and to advance genetic research in diverse populations. Real-world challenges drive the majority of methodology projects. The lab works closely with several large genetic consortia, including the Breast Cancer Association Consortium (BCAC), the Polygenic Risk Method in Diverse Populations Consortium (PRIMED), and the International Lung Cancer Consortium (ILCCO). I am looking for postdocs and students to work on fascinating research topics. If you are interested, please email me.
I received postdoc training in the Department of Biostatistics at Harvard T.H. Chan School of Public Health working with Xihong Lin. I received my Ph.D. from the Department of Biostatistics at Johns Hopkins Bloomberg School of Public Health under the guidance of Nilanjan Chatterjee and Ni Zhao. I also received the graduate student fellowship from National Cancer Institute under the guidance of Montserrat García-Closas.
- Statistical Genetics
- Risk Prediction
- Causal Inference
- Machine Learning
-
PhD in Biostatistics, 2019
Johns Hopkins University
-
BS in Statistics, 2014
Zhejiang University
Overview of Research
The current large data era provides unprecedented opportunities to understand human diseases, identify drug targets and develop personalized therapies. I am very excited to work on cutting-edge statistical methodologies and software for all kinds of data-driven problems to improve human health. My primary goal is to become a leading interdisciplinary researcher studying diverse populations and reducing health disparities in genetic research, with expertise in scalable statistical methods for analyzing multi-ethnic genetic and biobank data, advanced knowledge of underlying genetic architectures of complex traits and diseases.
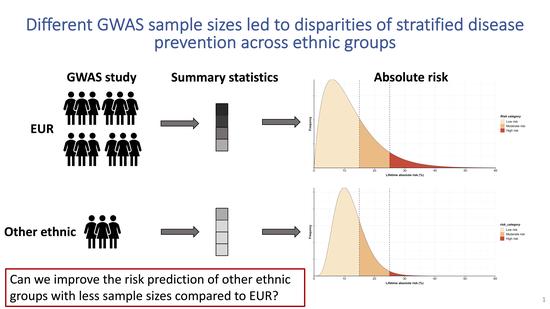
Multi-anestry polygenic risk predictions
Polygenic risk scores are useful for predicting various phenotypes; however, most PRS are developed using predominately European ancestry data, their performance in non-European populations is often poorer. To improve PRS performance in non-European populations, we propose a new method, which takes advantage of both existing large GWAS from European populations and smaller GWAS from non-European populations.
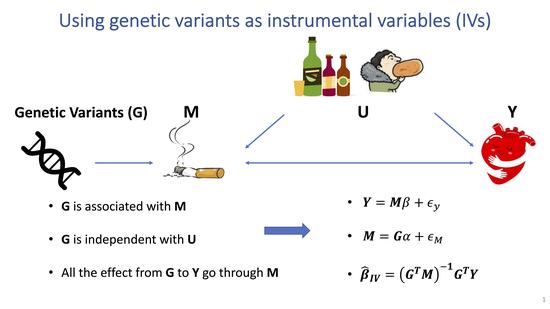
Robust Mendelian randomization methods incorporating weak and correlated instruments
Mendelian randomization (MR) is a major tool to test the causal association between risk factors and disease using genetic variants as instrumental variables.MR makes several strong assumptions, which can be violated in practice and lead to biased estimates and statistical inference. We develop a robust and powerful MR method requiring weaker and more realistic assumptions.
Testing for genetic association association and building risk prediction models for cancer incorporating tumor characteristics
Breast cancer represents a heterogeneous group of diseases with different molecular and clinical features. Thus, clarifying potential heterogeneous associations between genes and disease subtypes offers a tremendous opportunity to characterize distinct etiological pathways. I proposed several new computational and statistical approaches to develop powerful genetic associations tests.

Scientific collaborations
As a statistician and data scientist, I sincerely believe collaborations across disciplines are critical to scientific discovery and methodology development. I participated in broad research collaborations, including genetic association analysis for gall bladder cancer, effect-size distribution analysis for fourteen cancers, gene-environment interaction testing, risk factor analysis for bladder cancer, etc.
Featured Publication
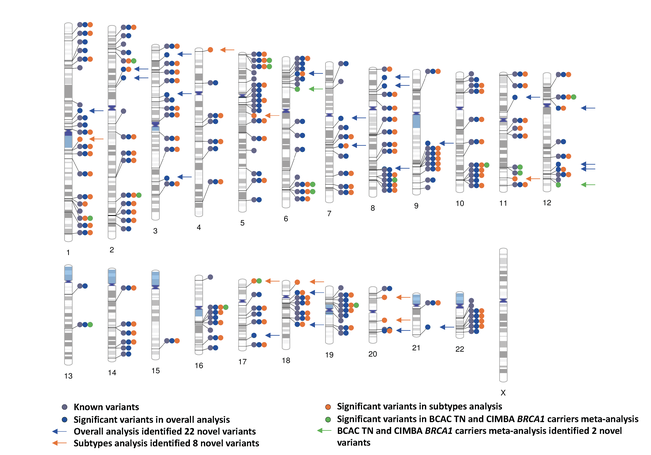
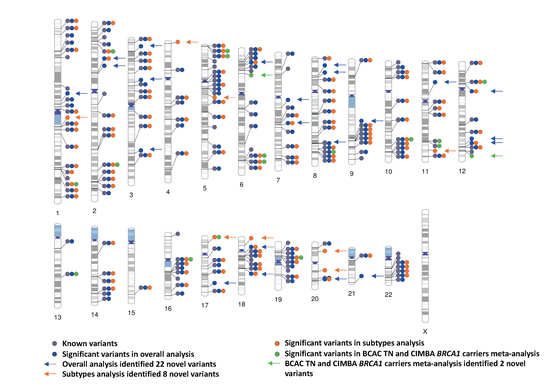
Genome-wide association study identifies 32 novel breast cancer susceptibility loci from overall and subtype-specific analyses
Breast cancer susceptibility variants frequently show heterogeneity in associations by tumor subtype. To identify novel loci, we performed a genome-wide association study including 133,384 breast cancer cases and 113,789 controls, plus 18,908 BRCA1 mutation carriers (9,414 with breast cancer) of European ancestry, using both standard and novel methodologies that account for underlying tumor heterogeneity by estrogen receptor, progesterone receptor and human epidermal growth factor receptor 2 status and tumor grade. We identified 32 novel susceptibility loci (P < 5.0 × 10−8), 15 of which showed evidence for associations with at least one tumor feature (false discovery rate < 0.05). Five loci showed associations (P < 0.05) in opposite directions between luminal and non-luminal subtypes.
Publications
Software

Talks & Posters
Teaching
Grants & Fellowship
Experience
Hobby
Aim to qualify for Boston marathon in 2025
Fan of Miami Heat!
Monster Hunter World, Civiliation 6, NBA 2K
Member of dragonboat team
Texas Holdem, Shengji, Mahjong