Assessment of polygenic architecture and risk prediction based on common variants across fourteen cancers
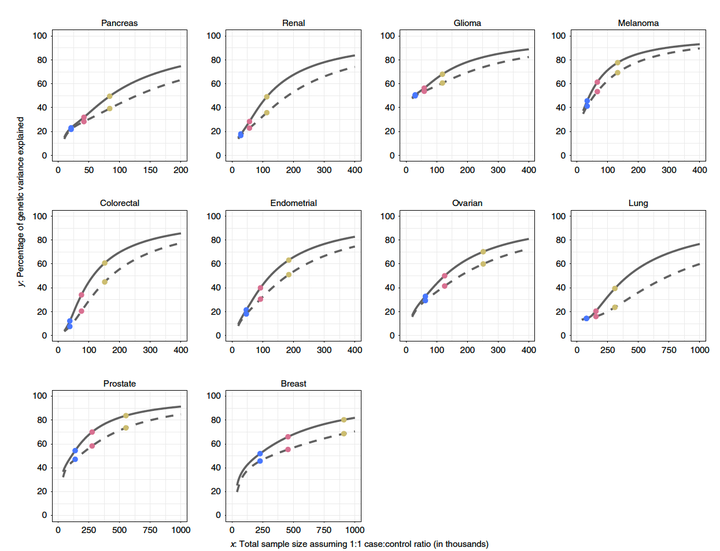 Projections of percentage of GWAS heritability explained by SNPs
Projections of percentage of GWAS heritability explained by SNPs
Abstract
Genome-wide association studies (GWAS) have led to the identification of hundreds of susceptibility loci across cancers, but the impact of further studies remains uncertain. Here we analyse summary-level data from GWAS of European ancestry across fourteen cancer sites to estimate the number of common susceptibility variants (polygenicity) and underlying effect-size distribution. All cancers show a high degree of polygenicity, involving at a minimum of thousands of loci. We project that sample sizes required to explain 80% of GWAS heritability vary from 60,000 cases for testicular to over 1,000,000 cases for lung cancer. The maximum relative risk achievable for subjects at the 99th risk percentile of underlying polygenic risk scores (PRS), compared to average risk, ranges from 12 for testicular to 2.5 for ovarian cancer. We show that PRS have potential for risk stratification for cancers of breast, colon and prostate, but less so for others because of modest heritability and lower incidence.