Power Analysis for Genetic Association Test (PAGEANT) provides insights to challenges for rare variant association studies
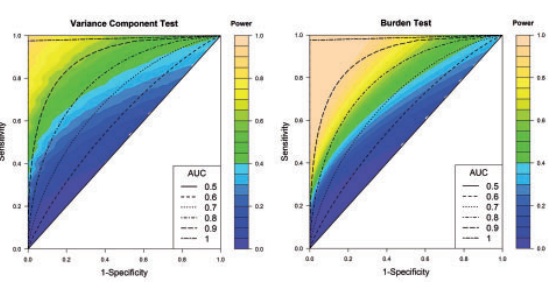 Effect of sensitivity and specificity for aggregate-level tests
Effect of sensitivity and specificity for aggregate-level tests
Abstract
We propose a new method for power calculation of aggregate level association rare variants tests. Compared to traditional power cacualtion, the new methods require a small number of key paprameters. We develop an analytic framework to obtain bounds on genetic architecture of an underlying trait given results from genome-wide association studies with rare variants. Finally, we provide insights into the required quality of annotation/functional information for identification of likely causal variants to make meaningful improvement in power.
Type
Publication
Bioinformatics